Sickle Cell Anemia : Understanding the Medical Complexity of a Hereditary Blood disorder
Introduction:

Sickle Cell Anemia is a hereditary blood disorder characterized by abnormal hemoglobin, leading to the production of sickle-shaped red blood cells. This condition affects millions of people worldwide, particularly those of African, Mediterranean, and Middle Eastern descent. In this blog, we will explore the medical aspects of Sickle Cell Anemia, including its etiology, clinical manifestations, diagnosis, and management. By gaining a deeper understanding of this condition from a medical professional perspective, we can provide comprehensive care and support to individuals living with Sickle Cell Anemia.

Etiology:
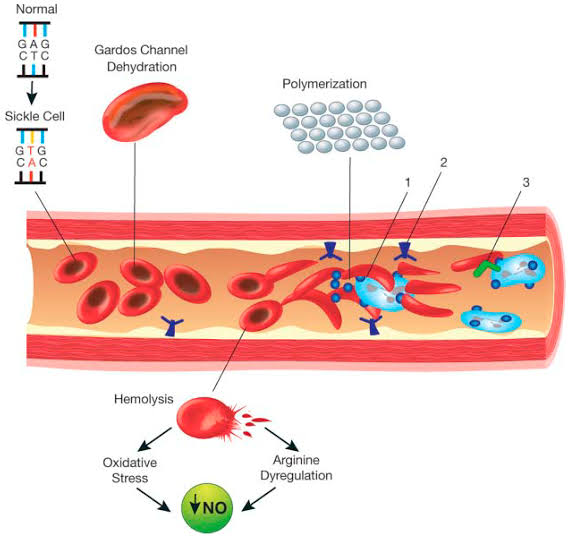
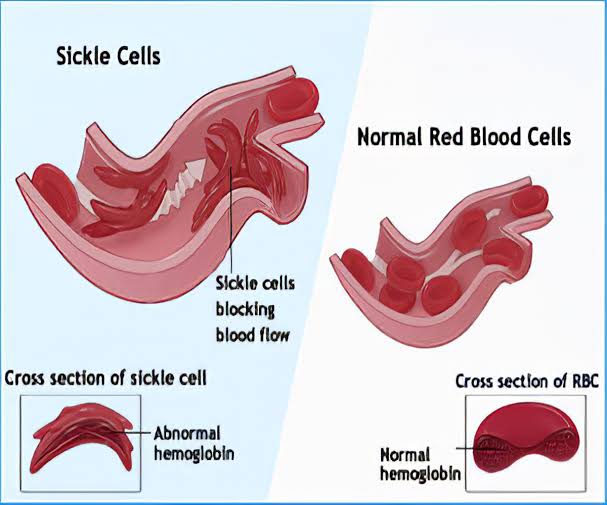
Sickle Cell Anemia is caused by a mutation in the gene that codes for hemoglobin, the protein responsible for carrying oxygen in red blood cells. The most common type of mutation is a single nucleotide substitution in which the amino acid valine replaces glutamic acid in the beta-globin chain. This mutation results in the production of abnormal hemoglobin called hemoglobin S (HbS). When deoxygenated, HbS causes red blood cells to become stiff and sickle-shaped, impairing their ability to flow smoothly through blood vessels.
Clinical Manifestations:
The clinical manifestations of Sickle Cell Anemia can vary widely among individuals and over time. The hallmark features include recurrent episodes of pain, anemia, and organ damage. The sickling of red blood cells can cause blockages in small blood vessels, leading to tissue ischemia (lack of oxygen) and acute painful episodes known as sickle cell crises. These crises can occur in various parts of the body and often require hospitalization and intensive pain management.
Chronic anemia is a common feature of Sickle Cell Anemia due to the destruction of sickle cells and the shortened lifespan of red blood cells. Anemia can cause fatigue, weakness, and shortness of breath.
Organ damage is another significant complication of Sickle Cell Anemia. The repeated episodes of blockage and ischemia can lead to damage in various organs, including the lungs, kidneys, liver, spleen, and bones. Chronic complications may include pulmonary hypertension, kidney dysfunction, liver cirrhosis, gallstones, and avascular necrosis of the bones.

Diagnosis:
Diagnosing Sickle Cell Anemia involves a combination of clinical evaluation, laboratory tests, and genetic testing. A complete blood count (CBC) reveals the presence of anemia, and a blood smear may show the characteristic sickle-shaped red blood cells. Hemoglobin electrophoresis is the definitive test for diagnosing Sickle Cell Anemia, identifying the abnormal hemoglobin S.

Prenatal testing, such as chorionic villus sampling or amniocentesis, can be performed to diagnose Sickle Cell Anemia in the fetus if there is a family history or if both parents are carriers.
Management and Support
The management of Sickle Cell Anemia aims to prevent complications, manage symptoms, and improve overall quality of life. This requires a multidisciplinary approach involving hematologists, pediatricians, internists, pain specialists, nurses, and other healthcare professionals.
Hydroxyurea is a medication commonly used in the management of Sickle Cell Anemia. It increases the production of fetal hemoglobin, which helps prevent sickling and reduces the frequency of painful crises. Blood transfusions may also be necessary in certain cases to manage severe anemia or acute complications.

Pain management is a critical aspect of care for individuals with Sickle Cell Anemia. The management of acute sickle cell crises typically involves the use of analgesics, hydration, and supportive care. Chronic pain management strategies may include the use of nonsteroidal anti-inflammatory drugs (NSAIDs), opioids, and non-pharmacological approaches such as physical therapy, heat therapy, and relaxation techniques.

To prevent complications, individuals with Sickle Cell Anemia require regular medical follow-up, vaccinations, and screenings for potential complications such as infections, organ damage, and stroke. Folic acid supplementation is often prescribed to support red blood cell production.
Psychosocial support and counseling are essential components of care for individuals with Sickle Cell Anemia and their families. Living with a chronic condition can be emotionally challenging, and individuals may benefit from education, coping strategies, and access to support groups.
Conclusion:
Sickle Cell Anemia is a complex hereditary blood disorder characterized by abnormal hemoglobin and sickle-shaped red blood cells. Understanding the medical aspects of Sickle Cell Anemia, including its etiology, clinical manifestations, diagnosis, and management, allows healthcare professionals to provide comprehensive care and support. Through a multidisciplinary approach, including medical interventions, pain management, preventive strategies, and psychosocial support, individuals with Sickle Cell Anemia can lead fulfilling lives while effectively managing the complications associated with this condition. Continued research, improved access to care, and public awareness are essential in advancing the management and outcomes for individuals affected by Sickle Cell Anemia



Comments
Post a Comment